The Data Science Garage
Using Data Science to Fight Cancer
The Lab
Our lab works on the intersection of computer engineering, statistical analysis and biology science. Based out of the OHSU Knight Cancer Institute we study systems biology, cancer, computational systems and integrative analysis. We work on projects funded by the Nation Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI).
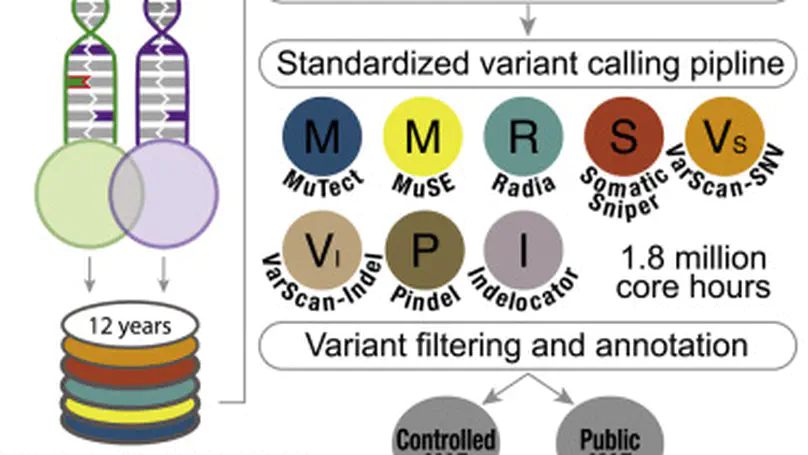
Projects








People
Researchers
Brian Walsh
Senior Research Software Engineer
Graph Databases, Machine Learning, Pan Cancer, Cross Project Data Harmonization
Quinn Wai Wong
Research Software Engineer
Software Development, Systems Design, Computational Biology
Grad Students
Principal Investigators
Kyle Ellrott
Associate Professor
Computational Biology, Data Science, Machine Learning, Precision Medicine, Cancer Early Detection